青海优质一次性引流袋厂家
发布时间:2022-11-26 00:40:19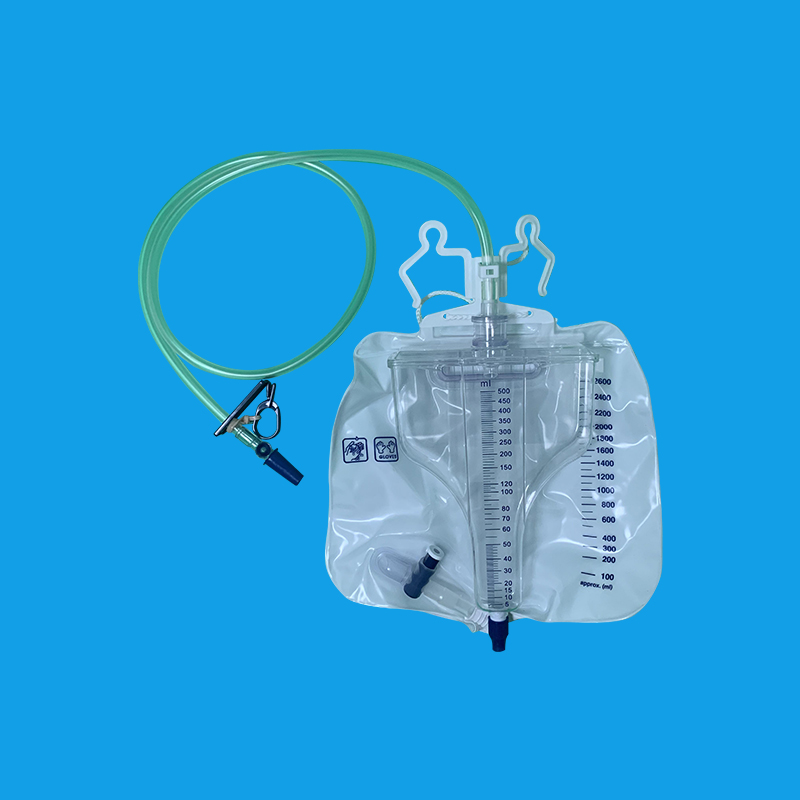
青海优质一次性引流袋厂家
二、明确要求建立鼓励创新医疗器械机制 新《条例》修订的核心内容之一是实行医疗器械注册人、备案人制度。实行医疗器械注册人、备案人制度,有利于鼓励产品创新、优化资源配置、落实主体责任、推动管理创新。 首先,新《条例》提出,“对创新医疗器械予以优先审评审批”,这就要求药品监管部门要建立健全创新医疗器械审评审批机制,强化审评员队伍建设,进一步优化审评流程,提高审评效率。 其次,新《条例》新增了未在境外上市的创新医疗器械,可以不提交(备案人)注册申请人所在国(地区)主管部门准许该医疗器械上市销售的证明文件。这将鼓励国际创新医疗器械首先在我国申报上市。 后,新《条例》新增对用于治疗罕见疾病、严重危及生命且尚无有效治疗手段的疾病和应对公共卫生事件等急需的医疗器械,可以作出附条件批准决定。明确了鼓励创新的方向,有利于加快创新产品上市。 广东省药品监管部门将以新《条例》颁布实施为契机,全面总结医疗器械注册人制度试点工作,坚持以创新发展为导向,坚持创新服务发展,助推创新产品上市,助力产业创新发展。研究出台深化医疗器械审批制度改革的政策举措,进一步优化审评审批流程,完善制度建设,提高审评审批效能。加强信息化建设,逐步将注册管理、生产许可、监督检查、监督抽检等信息有机整合,实现全省系统信息共享。加快推进医疗器械标识实施,深化卫健、医保、药监三医联动机制,全面推进药品监管体系和监管能力现代化综合改革。

青海优质一次性引流袋厂家
制备材料 1. 聚氯乙烯(PVC):产品较硬,刺激性较大,异物感强烈,价格低廉,多为无气囊产品。 2. 干胶(生胶):产品较软,刺激性较大,长时间使用容易导致导尿粘膜发炎,价格低廉,多为无气囊产品。 3. 硅橡胶:产品较软,生物相溶性好,病人无异物感,但成型过程表面不易保持光洁,成本高,多为无气囊产品。 4. 天然胶乳:产品柔软,生物相溶性好,病人感觉舒适,表面光洁度高,刺激性很小,价格适宜,多为气囊产品,留置操作方便。

青海优质一次性引流袋厂家
留置导尿管在临床上是很常见的一种操作,留置导尿管不仅可以缓解急性尿潴留病人腹部胀痛的情况,同时对于全麻手术的病人,通常也在术前需要留置导尿管。以男性病人为例,简单介绍留置导尿管的过程,首先患者要采取平卧位,操作前要对男性龟头以及尿道口进行充分消毒,消毒完毕以后铺无菌的操作单。其次打开无菌导尿包,用石蜡油将尿管润滑,左手将男性的阴茎拎起,右手用镊子夹住导尿管远端,插入男性尿道口,在插入的过程中要缓慢,避免对尿道黏膜造成损伤,看到尿液从尿管中流出时说明已经插入膀胱,用注射器打上球囊,就可以固定导尿管,导尿管远端接上引流袋就可以了。

青海优质一次性引流袋厂家
第四十条 从事医疗器械经营活动,应当有与经营规模和经营范围相适应的经营场所和贮存条件,以及与经营的医疗器械相适应的质量管理制度和质量管理机构或者人员。 第四十一条 从事第二类医疗器械经营的,由经营企业向所在地设区的市级人民政府负责药品监督管理的部门备案并提交符合本条例第四十条规定条件的有关资料。 按照国务院药品监督管理部门的规定,对产品安全性、有效性不受流通过程影响的第二类医疗器械,可以免于经营备案。 第四十二条从事第三类医疗器械经营的,经营企业应当向所在地设区的市级人民政府负责药品监督管理的部门申请经营许可并提交符合本条例第四十条规定条件的有关资料。 受理经营许可申请的负责药品监督管理的部门应当对申请资料进行审查,必要时组织核查,并自受理申请之日起20个工作日内作出决定。对符合规定条件的,准予许可并发给医疗器械经营许可证;对不符合规定条件的,不予许可并书面说明理由。 医疗器械经营许可证有效期为5年。有效期届满需要延续的,依照有关行政许可的法律规定办理延续手续。 第四十三条 医疗器械注册人、备案人经营其注册、备案的医疗器械,无需办理医疗器械经营许可或者备案,但应当符合本条例规定的经营条件。 第四十四条 从事医疗器械经营,应当依照法律法规和国务院药品监督管理部门制定的医疗器械经营质量管理规范的要求,建立健全与所经营医疗器械相适应的质量管理体系并保证其有效运行。 第四十五条 医疗器械经营企业、使用单位应当从具备合法资质的医疗器械注册人、备案人、生产经营企业购进医疗器械。购进医疗器械时,应当查验供货者的资质和医疗器械的合格证明文件,建立进货查验记录制度。从事第二类、第三类医疗器械批发业务以及第三类医疗器械零售业务的经营企业,还应当建立销售记录制度。
青海优质一次性引流袋厂家
加拿大CMDCAS 图片 加拿大医疗器械准入认证为CMDCAS认证,由卫生部发布,加拿大实行政府注册结合第三方的质量体系审查。这里所说的第三方,指经加拿大医疗器械认证认可机构(CMDCAS)认可的第三方机构。 分类 依据CMDR(加拿大医疗设备管理条例 ) SOR/98-282,根据器械的使用风险将医疗器械分为I、 II、III、IV四个分类,如I类器械为低风险,IV类器械风险为高。为此针对制造者提出的产品注册要求也是逐级增加,要求制造者实行的体系是愈加详尽。 注册流程 Class I: 1、为申请 MDEL(加拿大医疗器械营业许可证)准备相应的技术文件; 2、提交MDEL申请,支付卫生部行政收费; 3、申请评审通过,将在Health Canada网站公示。 Class II、III、IV: 1、通过CMDCAS认可的认证机构进行ISO 13485 审核认证(体系审核除ISO13485要求外还要包括CMDR的特殊要求),获得证书; 2、准备MDL(加拿大医疗器械许可证)申请; 3、提交MDL申请,并交纳卫生部行政收费; 4、Health Canada评审MDL申请, 评审通过后进行网站公示。 此外,加拿大对III、IV类医疗器械还需审核Premarket review documents(上市前审查文件)。 韩国KFDA 图片 韩国卫生福利部,简称卫生部,主要负责管食品、药品、化妆品和医疗器械的管理,是主要的卫生保健部门。 依照《医疗器械法》,韩国卫生福利部下属的食品药品安全部负责对医疗器械的监管工作。 韩国医疗器械法把医疗器械分为4类(Ⅰ、Ⅱ、Ⅲ、Ⅳ),从Ⅰ类至Ⅳ类风险程度递增,即:Ⅰ类为几乎没有潜在危险的医疗器械,Ⅳ类为高风险的医疗器械。 分类 Ⅰ类:几乎没有潜在危险的医疗器械; Ⅱ类:具有低潜在危险的医疗器械; Ⅲ类:具有中度潜在危险的医疗器械; Ⅳ类:高风险的医疗器械。 注册流程 1、确定产品分类(I、II、III、IV),选择韩代KLH(韩国证书持证人,不在韩国境内的企业需要选择一个韩国证书持证人,通常为在韩国的分销商); 2、II类产品需申请KGMP证书和接受现场审核,II类产品一般是授权的第三方审核员,并获得KGMP证书; 3、II类产品需要送样品到韩国MFDS授权的实验室进行韩国标准的测试; 4、由韩代向MFDS提交技术文件(检测报告,KGMP证书等),进行注册审批; 5、支付申请费用; 6、注册文件整改,注册批准; 7、指定韩国代理商和经销商,产品销售。