银川优质导尿回流袋批发
发布时间:2023-04-08 00:38:07
银川优质导尿回流袋批发
加拿大CMDCAS 图片 加拿大医疗器械准入认证为CMDCAS认证,由卫生部发布,加拿大实行政府注册结合第三方的质量体系审查。这里所说的第三方,指经加拿大医疗器械认证认可机构(CMDCAS)认可的第三方机构。 分类 依据CMDR(加拿大医疗设备管理条例 ) SOR/98-282,根据器械的使用风险将医疗器械分为I、 II、III、IV四个分类,如I类器械为低风险,IV类器械风险为高。为此针对制造者提出的产品注册要求也是逐级增加,要求制造者实行的体系是愈加详尽。 注册流程 Class I: 1、为申请 MDEL(加拿大医疗器械营业许可证)准备相应的技术文件; 2、提交MDEL申请,支付卫生部行政收费; 3、申请评审通过,将在Health Canada网站公示。 Class II、III、IV: 1、通过CMDCAS认可的认证机构进行ISO 13485 审核认证(体系审核除ISO13485要求外还要包括CMDR的特殊要求),获得证书; 2、准备MDL(加拿大医疗器械许可证)申请; 3、提交MDL申请,并交纳卫生部行政收费; 4、Health Canada评审MDL申请, 评审通过后进行网站公示。 此外,加拿大对III、IV类医疗器械还需审核Premarket review documents(上市前审查文件)。 韩国KFDA 图片 韩国卫生福利部,简称卫生部,主要负责管食品、药品、化妆品和医疗器械的管理,是主要的卫生保健部门。 依照《医疗器械法》,韩国卫生福利部下属的食品药品安全部负责对医疗器械的监管工作。 韩国医疗器械法把医疗器械分为4类(Ⅰ、Ⅱ、Ⅲ、Ⅳ),从Ⅰ类至Ⅳ类风险程度递增,即:Ⅰ类为几乎没有潜在危险的医疗器械,Ⅳ类为高风险的医疗器械。 分类 Ⅰ类:几乎没有潜在危险的医疗器械; Ⅱ类:具有低潜在危险的医疗器械; Ⅲ类:具有中度潜在危险的医疗器械; Ⅳ类:高风险的医疗器械。 注册流程 1、确定产品分类(I、II、III、IV),选择韩代KLH(韩国证书持证人,不在韩国境内的企业需要选择一个韩国证书持证人,通常为在韩国的分销商); 2、II类产品需申请KGMP证书和接受现场审核,II类产品一般是授权的第三方审核员,并获得KGMP证书; 3、II类产品需要送样品到韩国MFDS授权的实验室进行韩国标准的测试; 4、由韩代向MFDS提交技术文件(检测报告,KGMP证书等),进行注册审批; 5、支付申请费用; 6、注册文件整改,注册批准; 7、指定韩国代理商和经销商,产品销售。
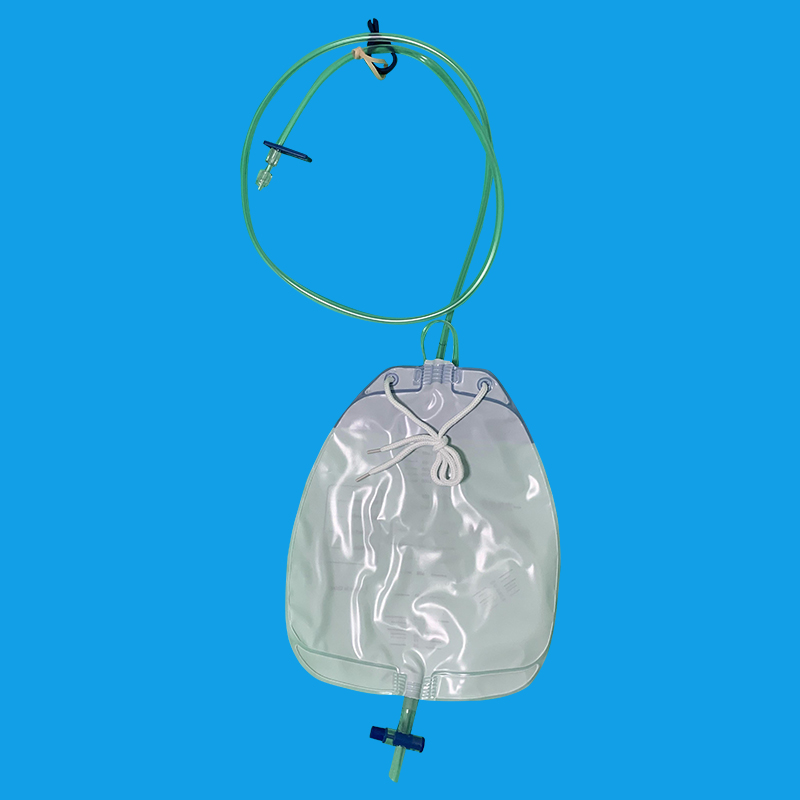
银川优质导尿回流袋批发
从全球市场规模来看,预计到2024年,病理学市场规模将从2019年的303亿美元达到444亿美元,从2019年到2024年复合年增长率为6.1%;从国内市场规模来看,病理行业的潜在市场超400亿元,其中组织病理市场规模20-30亿元、细胞病理学估计超300亿、免疫组化病理潜在市场空间超40亿、分子病理潜在检验空间超50亿元。 01 病理学分类 病理检查是肿瘤诊断的金标准。通过研究疾病的病因、发病机制、形态结构、功能和代谢等方面的改变,揭示疾病的发生发展规律,从而阐明疾病本质的医学科学。病理检查在肿瘤领域有着极为广泛的应用,虽然肿瘤的诊断有多种形式,但是病理报告目前被公认为是对肿瘤的“后判决”,是肿瘤诊断的“金标准”。 病理学根据发展阶段的不同主要分为传统病理技术(组织病理、细胞病理)、免疫组化病理和分子病理。传统的组织病理建立在组织、细胞的水平上,可以通过病理医师的显微镜诊断来判断疾病的性质(炎症性病变还是肿瘤型病变、良性肿瘤还是恶性肿瘤等);免疫组化病理建立在蛋白质水平上,可以进一步判断肿瘤的组织来源、原发部位、病理分型、残留边缘癌细胞等,除了诊断作用以外还具有指导预后的作用;分子病理建立在核酸分子水平上,可以确定肿瘤的基因突变类型,用于后续靶向药物的指导以实现精准医疗。 02 病理行业产业链 病理行业产业链包括上游原料市场、中游试剂设备市场和下游需求诊断市场。原料包括生物与化学原材料以及各种机械零配件,分子病理还会涉及到引物、探针等;中游包括病理诊断试剂和病理诊断仪器;下游需求主要来自于医院、第三方独立医学实验室、体检中心、政府等。 据2020年世界病理学大会报告,预计到2024年,病理学市场规模将从2019年的303亿美元达到444亿美元,从2019年到2024年的复合年增长率为6.1%。据GrandViewResearch预测,2019年全球数字病理学市场规模为7.676亿美元,预计到2027复合年增长率为11.8%。根据终用途,医院在2019年占据了领先的市场份额。医院采用数字扫描技术以加快诊断速度并提高患者依从性。就收入而言,2019年北美市场份额大。 病理AI属于AI+医疗领域的医学影像诊断细分领域,根据中国数字医疗网统计,2016年我国AI+医疗行业规模为96.61亿元,同比增长122.09%;2017年达130亿元,同比增长34.56%,2018年我国AI+医疗行业规模超200亿,同比增长53.85%。

银川优质导尿回流袋批发
(二)备案资料为鉴定依据 从案件行进时间来看,2017年3月30日,荣研化学在北京正式起诉广州迪澳;2018年12月13日,广州知识产权法院受理;2020年12月31日为一审判决日,整个案件从起诉到判决经历了5年多的时间。对比20年专利保护期,不难发现,一旦创新发明成果被侵权市场竞争将有失公平。 董巍介绍,本案件涉及的LAMP法两项专利中262已经过期,083在今年9月份也马上过期,“侵权者”正是抓住了这类案件取证难、判定难、耗时长、花费高等特点,才会表现出对专利保护敬畏度不够。一审判决出来后,被诉企业仍然在继续参与招投标,对判决结果可以说是置之不理。 他指出:“本案侵权专利为产品的一段人工引物设计,即便将涉嫌侵权的试剂盒交给鉴定机构,一般的鉴定机构在技术上也很难反向把引物从中成功分离出来,有很大的不确定性,带来原告举证不能的风险。冒着一审中因坚持不走鉴定途径进行技术比对而被驳回起诉的风险,我们仍然要求,调取该产品在国家药监局注册申请的备案资料进行技术对比。在原告的坚持下,一审法院采纳间接比对的方式并终认定被诉侵权产品落入了涉案两件发明专利权利要求保护范围,构成专利侵权。” 在国家大力鼓励产品创新的当下,加大专利保护是非常重要的一环。董巍呼吁,“要加快医疗器械注册审批专利链接制度落地,加大对恶意侵权行为单位及个人的惩罚力度,营造公平公正的国内外营商环境。” 《中华人民共和国专利法》(以下简称《专利法》)明确提出,对仿制药给予了一定的支持。在注册审批中引入专利审查,如果发现专利侵权纠纷就可通过法院裁定解决,把专利纠纷和侵权风险解决在产品上市之前。医疗器械类产品应该参照药品的该链接制度。 据悉,2004年荣研化学投资1020万美元,已在上海市张江高科技园生物医学园区设立了外商独资高科技生物医疗器械企业。荣研化学方表示,荣研化学早已确立了加快全球化企业建设的方针,高度重视中国市场,本案的胜诉坚定了进入中国市场的决心,他们将会把领先的技术、设备引入中国并进行推广。

银川优质导尿回流袋批发
得益于国家政策的扶持以及市场需求的增加,我国医疗器械行业销售规模近几年整体保持增长态势,医疗器械出口额度逐年增加:2019年出口总额达到891.29亿人民币,同比增长达13.13%;2020年医疗仪器及机械出口总额为1259.3亿人民币,同比增长41.5%。 但是,新版欧盟《医疗器械法规(MDR)》落地后,对国内所有医疗器械出口企业来说,都将产生翻天覆地的变化! 01 欧盟MDR法规,今日生效! 2017年5月5日,欧盟正式发布新版医疗器械法规MDR(EU 2017/745),取代旧的医疗器械指令MDD( 93/42/EEC)。新旧法规交替过渡期为三年。 2019年,海关总署组织技术性贸易措施研究评议基地成功应对欧盟MDR新规,促使欧盟在2020年4月24日正式宣布将《医疗器械法规(MDR)》强制实施日期推迟一年,这为中国医疗器械生产企业争取到了宝贵的缓冲时间,但强制执行日期为2021年5月26日,也就是昨天,《医疗器械法规(MDR)》正式落地! 02 新版法规,对械企的要求更高 根据欧盟新颁布的医疗器械法规《医疗器械法规》(2017/745,MDR)和《体外诊断器械法规》(2017/746,IVDR),欧盟将医疗器械分为两大类别:医疗器械MD和体外诊断器械IVD。 MDR法规执行时间为2021年5月26日,IVDR法规执行时间为2022年5月26日。所以,对于所有的体外诊断企业来说,还有一年的时间为欧盟的体外设备法规(IVDR)做准备。 据了解,MDR后侵入式医疗器械MD根据风险等级将再细分为 I、IIa、 IIb、III类;非侵入式体外诊断器械IVD依据风险等级由低到高细分为A、B、C、D四类。 总体来说,欧盟MDR新规更加关注临床性能、更好的医疗器械可追溯性和对患者更大的透明度,具体变化有:医疗器械的范围扩大;提出医疗器械新概念和定义;设立中央电子资料库(Eudamed);设立产品独立的产品识别码(UDI);完善了医疗器械的通用安全和性能要求;加强对技术文件的要求;加强器械上市后的监管;完善临床评价相关要求;对授权认证机构(NB)提出严格要求等。 这意味着对进入欧洲市场的医疗器械将实施更严格的限制,对医疗器械相关企业提出了更高的要求。
银川优质导尿回流袋批发
二、明确要求建立鼓励创新医疗器械机制 新《条例》修订的核心内容之一是实行医疗器械注册人、备案人制度。实行医疗器械注册人、备案人制度,有利于鼓励产品创新、优化资源配置、落实主体责任、推动管理创新。 首先,新《条例》提出,“对创新医疗器械予以优先审评审批”,这就要求药品监管部门要建立健全创新医疗器械审评审批机制,强化审评员队伍建设,进一步优化审评流程,提高审评效率。 其次,新《条例》新增了未在境外上市的创新医疗器械,可以不提交(备案人)注册申请人所在国(地区)主管部门准许该医疗器械上市销售的证明文件。这将鼓励国际创新医疗器械首先在我国申报上市。 后,新《条例》新增对用于治疗罕见疾病、严重危及生命且尚无有效治疗手段的疾病和应对公共卫生事件等急需的医疗器械,可以作出附条件批准决定。明确了鼓励创新的方向,有利于加快创新产品上市。 广东省药品监管部门将以新《条例》颁布实施为契机,全面总结医疗器械注册人制度试点工作,坚持以创新发展为导向,坚持创新服务发展,助推创新产品上市,助力产业创新发展。研究出台深化医疗器械审批制度改革的政策举措,进一步优化审评审批流程,完善制度建设,提高审评审批效能。加强信息化建设,逐步将注册管理、生产许可、监督检查、监督抽检等信息有机整合,实现全省系统信息共享。加快推进医疗器械标识实施,深化卫健、医保、药监三医联动机制,全面推进药品监管体系和监管能力现代化综合改革。