新密优质集尿袋批发
发布时间:2024-03-26 00:33:50
新密优质集尿袋批发
欧盟CE 欧盟为规范其市场,统一产品标准和规范,规定在欧盟市场“CE”标志属强制性认证标志,不论是欧盟内部企业还是其他国家生产的产品,要想在欧盟市场上自由流通,就必须加贴“CE”标志,以表明产品符合欧盟《技术协调与标准化新方法》指令的基本要求。这是欧盟法律对产品提出的一种强制性要求,出口到欧盟的医疗器械没有CE认证无法清关。 图片 欧盟CE 认证标志 分类 根据欧盟新颁布的医疗器械法规《医疗器械法规》(2017/745,MDR)和《体外诊断器械法规》(2017/746,IVDR),现在欧盟范围内医疗器械只分两类:医疗器械MD和体外诊断器械IVD。 根据申明的产品预期使用目的,如果产品是侵入式的或接触到皮肤的,则属于MDR管辖的医疗器械MD;如果是非侵入式的或完全接触不到皮肤的,则属于IVDR管辖的体外诊断器械IVD。 根据《医疗器械法规》(MDR),医疗器械MD根据风险等级分为 I、IIa、 IIb、III类。 而根据《体外诊断器械法规》(IVDR),体外诊断器械IVD依据风险等级由低到高分为A、B、C、D四类。 新规主要变化 一是在法规层级上将指令上升为法规; 二是该两个医疗器械法规取代了之前的三个医疗器械指令; 三是体外诊断器械分类规则变化较大。 图片 2017年欧盟颁布的两个新法规2017年生效,但有一定过渡期,分别于2021年5月和2022年5月强制实施。 CE认证流程 澳大利亚TGA 澳大利亚药品管理局(TGA),是澳洲医疗用品的监管机构,负责一系列评估和监管确保澳洲药品保质保量。TGA监管的产品范围包括药品,医疗器械,血液及血液产品。 分类 澳大利亚将医疗器械根据其构成的风险程度分为五大类。分类级别越高,要求就越苛刻。 准入程序 一、如果医疗器械是在海外制造的(即I类消毒; I类测量; IIa类; IIb类,III类,AIMD类) 1. 制造商从TGA或欧盟的认证机构获得合格评定证据; 2. 制造商准备澳大利亚符合性声明; 3. 主办者向TGA提交制造商的证据; 4. 主办者递交在ARTG登记申请; 5. 在ARTG登记后主办者可以在澳大利亚供应器械; 6. 器械上市后持续监控。 二、如果该器械包含一种药物或动物材料,微生物或人类起源物(即 III类和 AIMD类): 1. 制造商决定质量规程,用于证示器械符合相关的基本原则,并准备必要的文件; 2. 制造商申请TGA合格评定证; 3. 制造商准备澳大利亚符合性声明; 4. 主办者向TGA提交制造商的证据; 5. 主办者递交在ARTG登记申请; 6. 在ARTG登记后主办者可以在澳大利亚供应器械; 7. 器械上市后持续监视。

新密优质集尿袋批发
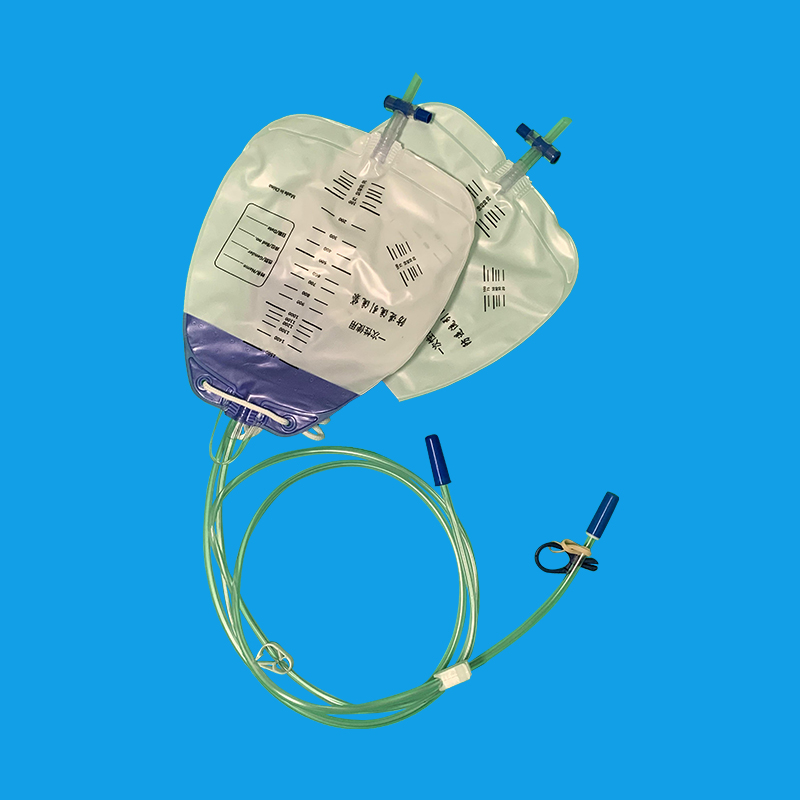
注意事项 1. 严格执行无菌操作:误插阴道或脱出立即更换。 2. 控制尿潴留患者放尿速度和量:勿快,放600-800ml夹管。 3. 观察记录尿颜色、量、性质 1) 正常:1500-2000ml/24h;多尿:>2500ml/24h;少尿:<400ml/24h;无:<50ml/24h。 2) 色:正常:无色透明或淡黄色;异常:血尿、血红蛋白尿、胆红素尿、乳糜尿。 3) 妥善固定尿管,保持管道通畅,堵塞时及时检查并调整尿管位置,用呋喃西林反复冲洗必要时更换。 4) 预防泌尿系统感染不必要每天进行膀胱冲洗,需尿道口擦洗二次,病情稳定后早拔管严格执行无菌操作,每日更换尿袋,长期留管者每周更换导尿管一次,留管期间鼓励患者多饮水。 5) 预防尿道出血渗尿:插入上尿管见尿再进4-5cm后充气或注水,然后将导尿管向外轻拉至不动即可,可有效预防尿道出血或渗液。

新密优质集尿袋批发
更重要的是,随着行业发展,医疗器械的经销商们已经在不知不觉间成为强势的一方,对生产商有了话语权。 医疗器械销售这一行是典型的“赢者通吃”,市场、渠道、关键性人物就那么几个(主要是当地联采办、三甲医院院长等),且不具备可替代性,因而医疗器械行业两类人具有优势:先发者,或者关系大户。 而地区的大经销商,基本上两类都是,早些年医疗器械跨国药企大举进入中国时,招聘总监级别的岗位时,没点政府背景都入不了他们的法眼。而这些老玩家们多年深耕经营,不仅积累了大量人脉和经验,也基本掌握了地区大部分的医疗器械代理渠道。 因此,即使新进入市场的企业有直销的念头,也很难突破藩篱、承担成本,终往往还是投向了经销商的怀抱。 这是另一层面上的垄断——经销商对销售渠道的垄断。技术壁垒和流通渠道,兜兜转转,价格虚高始终离不开“垄断”二字。 那么,如何才能让医疗器械的价格降下来? 如前文所述,医疗器械的高价格分为生产和流通两个部分。前者囿于海外药企的技术垄断所导致的高定价,下游自然没有话语权;而另一边,代理商们又可以仗着渠道优势肆无忌惮的加价。 医疗器械的高定价需要引入足够的玩家来用市场化机制破除;而集采大背景下,药品直接打包给医院,经销商可能会随着加价空间的消失,而逐渐退出历史舞台。 也就是说,集采既能够打破外企高价垄断,又能压缩经销商的多级抬价。这是近几年医改探索出来的控费之路,也要逐渐应用在医疗器械领域,而这一切的前提,就是细分领域有足够多的竞争玩家。 讲道理,进口医疗器械受限于本国高昂的研发、审批、劳动力和运输等成本,而国内企业基于模仿,研发、人力、原材料成本都相对低廉,又受到政策倾斜照顾,在成本方面具有不可比拟的优势。 然而现实情况是,因为起步时间晚,很多国产器械只能走低端化路线,但质量始终没能迎头赶上。所以,近几年国内医疗器械市场规模在不断扩大,而不良反应报告也逐年增多,直到2019年才有所好转。 这种情况下,消费者的选择也很真实。虽然国产设备厂商很愿意与医院方多做交流,在设备使用中,会根据院方的意见,在性能和设计上做进一步完善,遇到什么问题,会及时解决,医生操作起来更顺手;然而,当医院做一些科研或教学任务时,仍然会使用进口设备。 而另一种情况是,很多国产器械在走汽车行业的老路:不少国产CT机虽然是自己的,但其中诸如CT探测器(相当于照相机的感光元件)、信号链(处理图像的专用芯片模块)仍然绕不过前面提到的GPS三巨头。 国内的医疗器械虽然通过“连抄带借”完成了一部分自主化,但这终究只是企业短期生存的需要,而这种精细制造业是一个长期积累的过程,这二者之间的矛盾让本土的医疗器械直到如今也没能走出“低端制造”的怪圈。 我国医疗器械公司呈现小、散、乱、多的局面,我国有超过18000家医疗器械企业,其中90%以上规模在2000万元以下。 早在2015年,“生物医药及高性能医疗器械”就被列入《中国制造2025》,成为重点突破发展的十大领域之一,与信息技术、航空航天等核心产业并驾齐驱。 中美贸易摩擦开始后,特朗普用几道禁令轻而易举地掐住了华为、中兴的喉咙。从那时起,“自主可控”有一次成了我国高科技产业的一个执念。谈论多的当然是半导体。不过,疫情冲击下,也有越来越多人开始意识到在医药领域自主崛起的重要性。 如果说半导体行业关乎人的发展,那么医药行业关乎的是人的生存。民众被“看病难”“看病贵”“天价进口药”“天价手术”折磨的那些年,是时候成为历史了。 在药品领域,全国性集采已经常态化,在药品领域,当年的“神药”和“仿制药大户”已经逐渐在向创新药企的转型。而在“脱钩”之声此起彼伏的当下,IVD、彩超、CT、手术机器人等器械领域的突破自然也刻不容缓。中国高科技产业的攻坚,不能只有一个华为。
新密优质集尿袋批发
加拿大CMDCAS 图片 加拿大医疗器械准入认证为CMDCAS认证,由卫生部发布,加拿大实行政府注册结合第三方的质量体系审查。这里所说的第三方,指经加拿大医疗器械认证认可机构(CMDCAS)认可的第三方机构。 分类 依据CMDR(加拿大医疗设备管理条例 ) SOR/98-282,根据器械的使用风险将医疗器械分为I、 II、III、IV四个分类,如I类器械为低风险,IV类器械风险为高。为此针对制造者提出的产品注册要求也是逐级增加,要求制造者实行的体系是愈加详尽。 注册流程 Class I: 1、为申请 MDEL(加拿大医疗器械营业许可证)准备相应的技术文件; 2、提交MDEL申请,支付卫生部行政收费; 3、申请评审通过,将在Health Canada网站公示。 Class II、III、IV: 1、通过CMDCAS认可的认证机构进行ISO 13485 审核认证(体系审核除ISO13485要求外还要包括CMDR的特殊要求),获得证书; 2、准备MDL(加拿大医疗器械许可证)申请; 3、提交MDL申请,并交纳卫生部行政收费; 4、Health Canada评审MDL申请, 评审通过后进行网站公示。 此外,加拿大对III、IV类医疗器械还需审核Premarket review documents(上市前审查文件)。 韩国KFDA 图片 韩国卫生福利部,简称卫生部,主要负责管食品、药品、化妆品和医疗器械的管理,是主要的卫生保健部门。 依照《医疗器械法》,韩国卫生福利部下属的食品药品安全部负责对医疗器械的监管工作。 韩国医疗器械法把医疗器械分为4类(Ⅰ、Ⅱ、Ⅲ、Ⅳ),从Ⅰ类至Ⅳ类风险程度递增,即:Ⅰ类为几乎没有潜在危险的医疗器械,Ⅳ类为高风险的医疗器械。 分类 Ⅰ类:几乎没有潜在危险的医疗器械; Ⅱ类:具有低潜在危险的医疗器械; Ⅲ类:具有中度潜在危险的医疗器械; Ⅳ类:高风险的医疗器械。 注册流程 1、确定产品分类(I、II、III、IV),选择韩代KLH(韩国证书持证人,不在韩国境内的企业需要选择一个韩国证书持证人,通常为在韩国的分销商); 2、II类产品需申请KGMP证书和接受现场审核,II类产品一般是授权的第三方审核员,并获得KGMP证书; 3、II类产品需要送样品到韩国MFDS授权的实验室进行韩国标准的测试; 4、由韩代向MFDS提交技术文件(检测报告,KGMP证书等),进行注册审批; 5、支付申请费用; 6、注册文件整改,注册批准; 7、指定韩国代理商和经销商,产品销售。