石家庄优质导尿回流袋厂家
发布时间:2024-07-27 00:32:49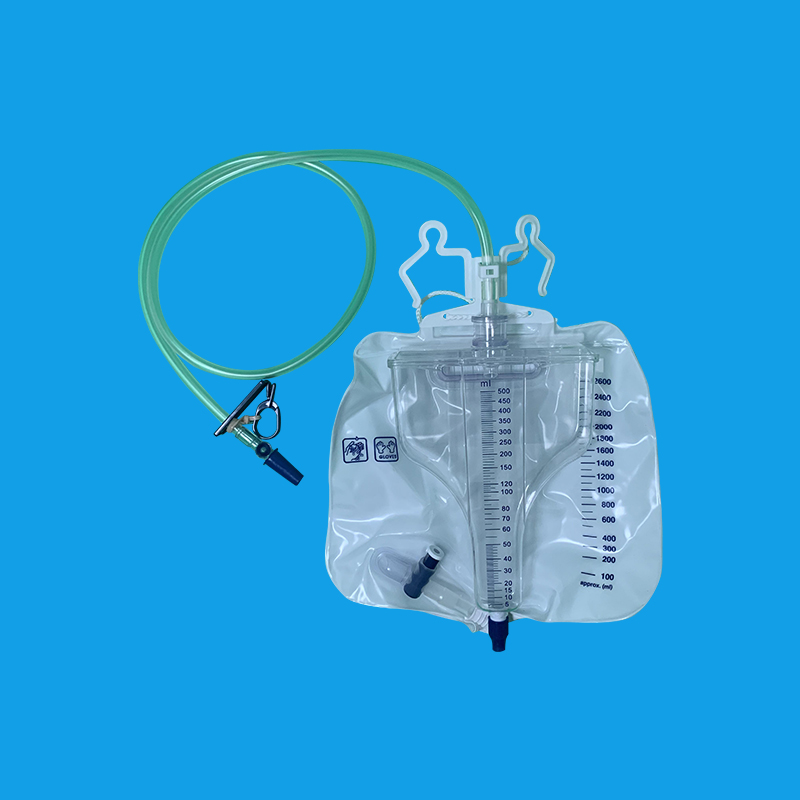
石家庄优质导尿回流袋厂家
导尿管护理步骤 1. 洗手、清除手上的病菌以预防感染。 2. 准备以下用具:棉签一包、透气纸胶布一卷、塑胶袋一只、便盆、生理食盐水或煮沸过的水、优碘消毒溶液(视需要)。 3. 将便盆放入病人臀部。 4. 用手分开阴唇或回缩包皮。 5. 以棉签沾湿生理食盐水或煮沸过的水,清洁靠近导尿管口端的导尿管约一寸(2.5公分),每次使用一枝棉签,将脏的棉签丢入准备好的塑胶袋内。 6. 检查有没有任何结痂或不正常引流物或分泌物。如果有,请告知医护人员处理。 7. 将导尿管用透气胶布以井字形贴法固定在大腿内侧(女病人)或下腹部(男病人),每天须更换黏贴部位,防止长期黏贴或导管压迫,造成皮肤损伤。

石家庄优质导尿回流袋厂家
欧盟CE 欧盟为规范其市场,统一产品标准和规范,规定在欧盟市场“CE”标志属强制性认证标志,不论是欧盟内部企业还是其他国家生产的产品,要想在欧盟市场上自由流通,就必须加贴“CE”标志,以表明产品符合欧盟《技术协调与标准化新方法》指令的基本要求。这是欧盟法律对产品提出的一种强制性要求,出口到欧盟的医疗器械没有CE认证无法清关。 图片 欧盟CE 认证标志 分类 根据欧盟新颁布的医疗器械法规《医疗器械法规》(2017/745,MDR)和《体外诊断器械法规》(2017/746,IVDR),现在欧盟范围内医疗器械只分两类:医疗器械MD和体外诊断器械IVD。 根据申明的产品预期使用目的,如果产品是侵入式的或接触到皮肤的,则属于MDR管辖的医疗器械MD;如果是非侵入式的或完全接触不到皮肤的,则属于IVDR管辖的体外诊断器械IVD。 根据《医疗器械法规》(MDR),医疗器械MD根据风险等级分为 I、IIa、 IIb、III类。 而根据《体外诊断器械法规》(IVDR),体外诊断器械IVD依据风险等级由低到高分为A、B、C、D四类。 新规主要变化 一是在法规层级上将指令上升为法规; 二是该两个医疗器械法规取代了之前的三个医疗器械指令; 三是体外诊断器械分类规则变化较大。 图片 2017年欧盟颁布的两个新法规2017年生效,但有一定过渡期,分别于2021年5月和2022年5月强制实施。 CE认证流程 澳大利亚TGA 澳大利亚药品管理局(TGA),是澳洲医疗用品的监管机构,负责一系列评估和监管确保澳洲药品保质保量。TGA监管的产品范围包括药品,医疗器械,血液及血液产品。 分类 澳大利亚将医疗器械根据其构成的风险程度分为五大类。分类级别越高,要求就越苛刻。 准入程序 一、如果医疗器械是在海外制造的(即I类消毒; I类测量; IIa类; IIb类,III类,AIMD类) 1. 制造商从TGA或欧盟的认证机构获得合格评定证据; 2. 制造商准备澳大利亚符合性声明; 3. 主办者向TGA提交制造商的证据; 4. 主办者递交在ARTG登记申请; 5. 在ARTG登记后主办者可以在澳大利亚供应器械; 6. 器械上市后持续监控。 二、如果该器械包含一种药物或动物材料,微生物或人类起源物(即 III类和 AIMD类): 1. 制造商决定质量规程,用于证示器械符合相关的基本原则,并准备必要的文件; 2. 制造商申请TGA合格评定证; 3. 制造商准备澳大利亚符合性声明; 4. 主办者向TGA提交制造商的证据; 5. 主办者递交在ARTG登记申请; 6. 在ARTG登记后主办者可以在澳大利亚供应器械; 7. 器械上市后持续监视。
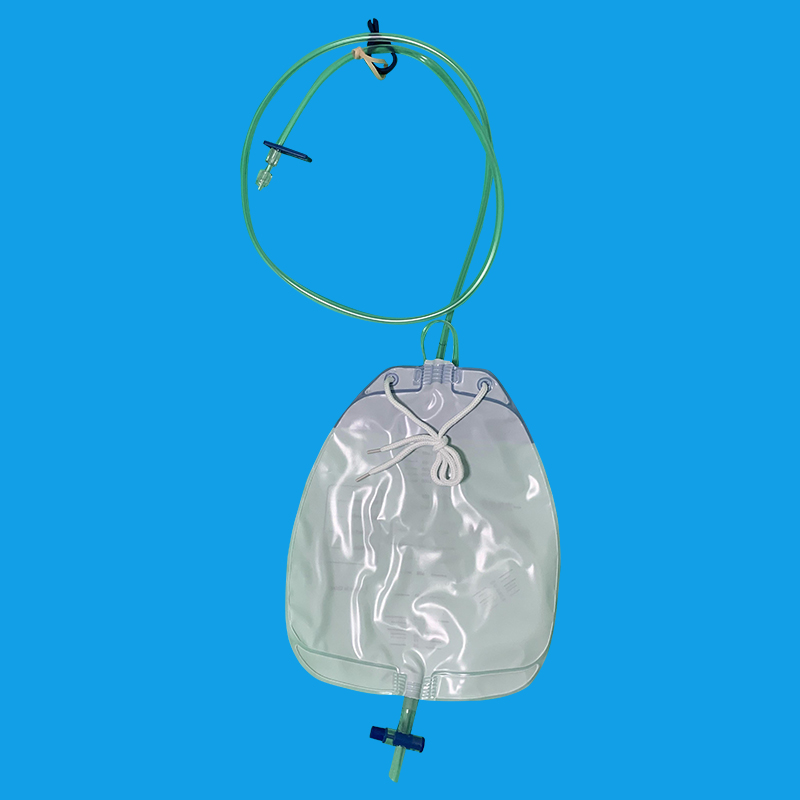
石家庄优质导尿回流袋厂家
医疗器械是与人类生命健康息息相关的产品,世界各国对医疗器械行业的发展高度重视。近年来随着我国经济发展“供给侧结构性改革”战略的实施,将医疗器械产业的发展列入了国家发展战略中,医疗器械行业进入高速发展期,逐渐跻身国际医疗器械行业,从消费大国逐渐转化为制造大国和出口大国。 随着全球公众健康意识增强,医疗服务机构对高品质医疗器械的要求不断提高,各国政府为提升医疗器械的安全性制定了相应的法律法规,以保护民众的生命安全。 因此我国医疗器械行业在走出国门时有必要对相关国家的法律法规有所了解,并知晓相应的监管要求和市场需求的方向,针对性地采取相应的策略和措施,稳步推进,不断提高产品的能级,逐步扩大海外市场。本期小编整理了中国主要出口国家和地区医疗器械的准入要求,供大家参考。 1、医疗器械定义 医疗器械是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件。 2.常见医疗器械产品海关税则号与名称 3、主要国家和地区分类及要求 美国FDA 美国食品药品监督管理局(简称:FDA),其职责是确保美国本国生产或进口的食品、化妆品、药物、生物制剂、医疗设备和放射产品等的安全,同时也负责执行公共卫生条件及州际旅行和运输的检查、对于诸多产品中可能存在的疾病的控制等。 分类 医疗器械范围很广,小到医用手套,大至心脏起博器,均在FDA监督之下,根据医疗用途和对人体可能的伤害,FDA将医疗器械分为Ⅰ、Ⅱ、Ⅲ类,等级越高监督越严。 一类(低至中度风险):普通管理。是指风险小或无风险的产品。 第二类(中度至高度风险):普通+特殊管理。其管理是在“普通管理”的基础上,还要通过实施标准管理或特殊管理,以保证质量和安全有效性。 第三类(高风险):上市前批准管理(PMA)。是指具有较高风险或危害性,或是支持或维护生命的产品,例如人工心脏瓣膜、心脏起搏器、人工晶体、人工血管等。 准入要求 对Ⅰ类产品,企业向FDA递交相关资料并审核通过后,FDA只进行公告,但并无相关证件发给企业; 对Ⅱ、Ⅲ类器械,企业须递交PMN或PMA,FDA在公告的同时,会给企业以正式的市场准入批准函件(Clearance),即允许企业以自己的名义在美国医疗器械市场上直接销售其产品。至于申请过程中是否到企业进行现场GMP考核,则由FDA根据产品风险等级、管理要求和市场反馈等综合因素决定。

石家庄优质导尿回流袋厂家
MDx组委: 您和团队在新冠疫情中也有非常多的工作与成果,那么也请您谈谈对于变异株频繁发现等全球疫情背景下,您对于新冠病毒检测的新思考?接下来应当引起注意的有哪些关键点?对于新冠疫苗,中和抗体检测方面您有哪些建议想法? 李教授: 变异株频现的背景下,新冠病毒检测试剂首先要自证清白,主要是漏检问题,包括灵敏度降低甚至假阴性问题,无论核酸检测还是抗原检测都需要拿出数据,无论真实世界数据还是模拟临床数据。新冠同多数疾病一样,会呈现出多种检测系统共存的局面,孰优孰劣,往往不是技术说了算,而是市场说了算。 疫苗接种后,需要关注的是抗原性漂移(drift)和转移(shift)问题,二者分属量变到质变不同阶段,但都需要密切监测变异的发生和累积,变异检测要常规开展起来。实际上,目前也是这么做的,但仅仅测序不够,还需要更高密度的人群监测,这需要快速检测试剂,尤其是针对那些值得警惕的变异(variant of concern, VOC)和值得关注的变异(variant of interest, VOI)。可以预期,VOC和VOI的名单会不断变化,需要人们快速行动,及时推出针对性的快速检测体系来。 MDx组委: 您在核酸检测,多重核酸分析等方向均有丰富的研究及转化成果,那么对于在新冠刺激下目前行业内风头正起的病原多重联合检测技术,想听听您的看法和评价如何,尤其是在结核等传染病的分子诊断方面,对于行业有什么建议? 李教授: 相对于新冠病毒而言,结核等其它传染病的分子诊断更为复杂:比如,多病原体联合检测就面临检测平台、临床试验和临床意义等多方面挑战。 就结核而言,分枝杆菌是一大类病原体,感染症状相似,但治疗手段差别很大,需要甄别出结核分枝杆菌和非结核分枝杆菌的具体种类,本身就极具挑战性。目前,我国结核分枝杆菌核酸筛查普及率不够高,一个特殊原因是合适的样本类型还未得到很好的解决。再有就是耐药突变的发生,这是结核治疗的老大难问题,耐药检测不充分,和治疗不充分一样,会导致大量问题的发生。 我国对于新冠确实控制得很好,目前是新冠肺炎低负担国家,然而不幸的是,我国一直都是结核病高负担国家,和印度、印尼等发展中国家一起名列前茅。原因固然很多,但与总体投入偏少不无关系。分子诊断行业固然需要继续努力,持续研发质优价廉的诊断产品,但疾病本身的防控也需要全社会的高度重视。 MDx组委: 您作为厦大分子诊断教育部工程研究中心主任以及致善生物董事长,也想请您简单谈谈,接下来在后疫情时代,您和厦大团队在分子诊断领域的研究计划,以及关于致善在分子诊断领域的布局有哪些思考? 李教授: 我们这个团队的底色决定了它的研究型和创新型特质。一方面,我们将继续深耕多靶标熔解曲线技术这一优势技术领域,通过持续的研发投入和技术创新,力争解决一些相对复杂的分子诊断难题;另一方面,也将围绕分子诊断的关键问题,通过多种形式的合作,努力打造良性的生态系统,形成多方共赢的局面。 MDx组委:感谢李教授的精彩分享!敬请期待李教授在会议现场带来更多专业分享!